|
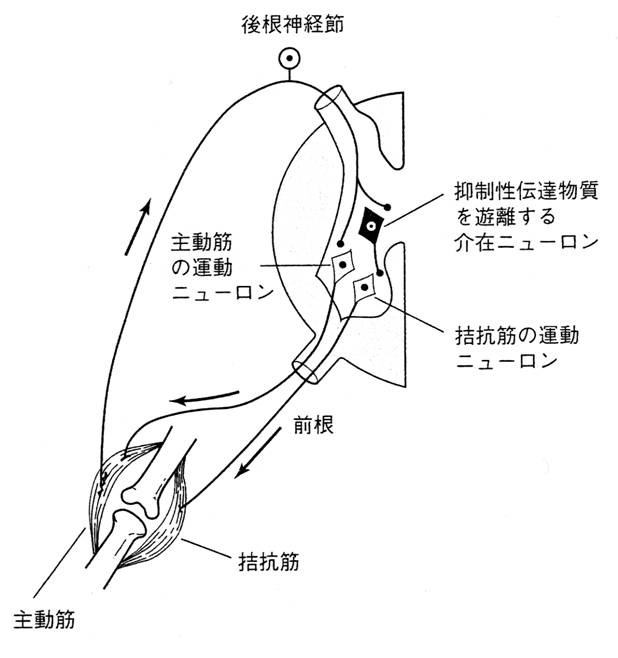
グリシンによるシナプス伝達とびっくり病での遺伝子変異 |
| |
(1) Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell, Wasmuth JJ. (1993) Mutations
in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant
neurologic disorder, hyperekplexia. Nat Genet. 5, 351-358
(2) Ryan SG,
Buckwalter MS, Lynch JW, Handford CA, Segura L, Shiang R, Wasmuth JJ, Camper
SA, Schofield P, & O'Connell P. (1994) A missense mutation in
the gene encoding the alpha 1 subunit of the inhibitory glycine receptor in the
spasmodic mouse.Nat Genet. 7, 131-135.
(3) Rees MI, Harvey K,
Pearce BR, Chung SK, Duguid IC, Thomas P, Beatty S, Graham GE, Armstrong L,
Shiang R, Abbott KJ, Zuberi SM, Stephenson JB, Owen MJ, Tijssen MA, van den
Maagdenberg AM, Smart TG, Supplisson S, & Harvey RJ.(2006) Mutations
in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human
startle disease. Nat Genet. 38, 801-806.
Note 1 Hyperekplexia
「驚愕反射の亢進」は症候論ではhyperekplexiaと呼ばれる。hyperexplexiaとの記載もあるが、原著によるとhyperekplexiaが正しい。
Note 2 予期しない刺激の処理
「筋緊張の相反性支配がうまくいかない」ことは、「手足が棒のようになる」ことを説明するが、「驚きやすい」ことを説明しない。予期しない刺激(音、触覚、なんでもよい)を処理する脳幹部のシステムにもグリシンが絡んでいるはず。グリシン受容体は、脳幹部から脊髄に発現しているが、それより上の中枢神経系では発現していない。
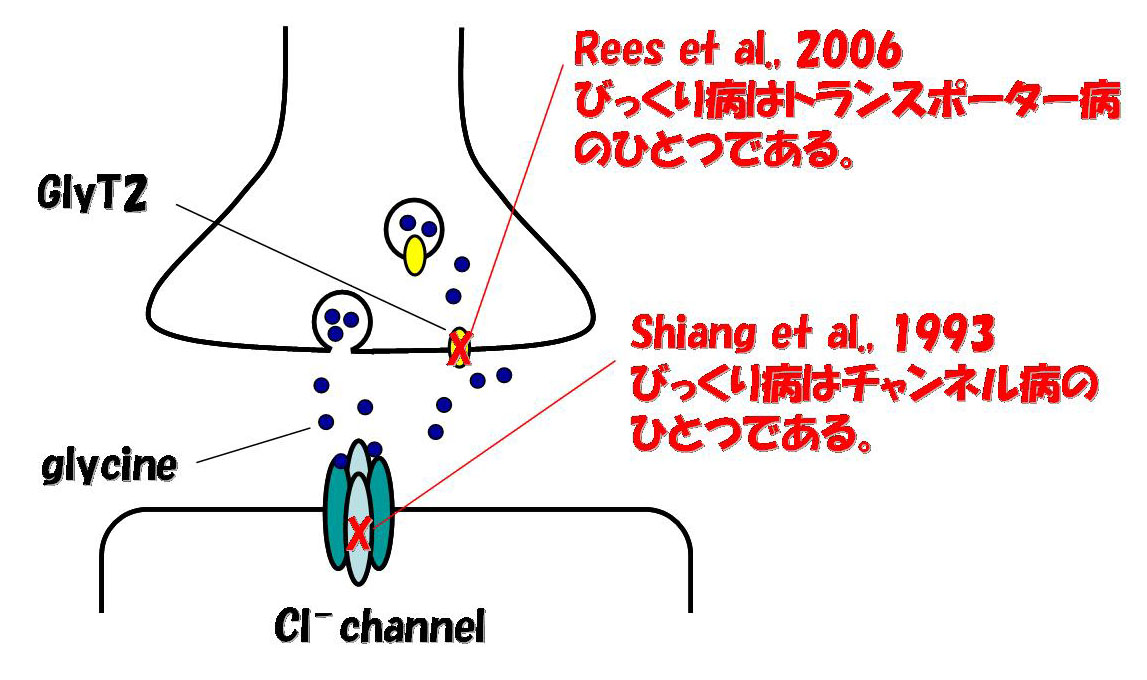
チャンネル病とトランスポーター病

生理の教科書に戻る。水、電解質などの極性のある低分子は脂質二重膜層を通過できず、それらを細胞内に入れる/細胞から排出するためには、膜に埋まったたんぱく質が必要である。相手にする分子、および性質の違いから、これらのたんぱく質はチャンネルとトランスポーターに大別される。
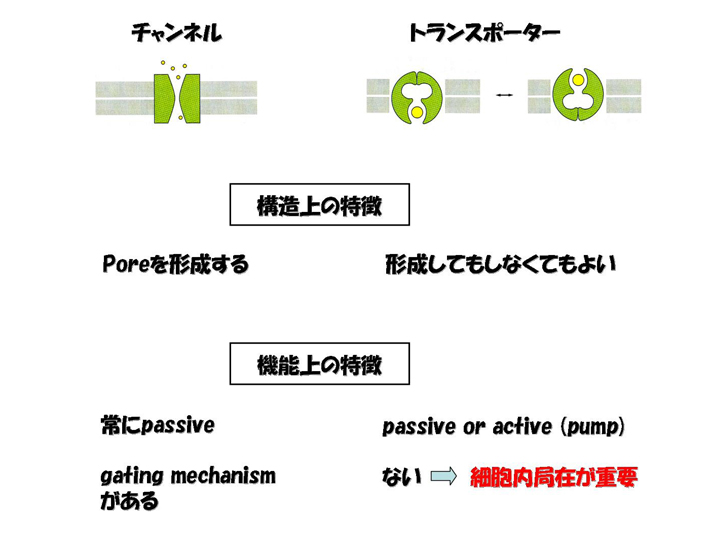
チャンネルが通すのは水または電解質である。その構造上の特徴として、必ずポアを形成し、開閉いずれかの状態をとる。分子の移動は濃度勾配に従い、エネルギーを必要としない(passiveである)。開閉を制御するメカニズム(gating
mechanism)がある。これに対して、トランスポーターは電解質、アミノ酸、糖、金属、薬物など、水以外のありとあらゆる低分子の輸送を担う。その構造、機能ともに、チャンネルに比べるとルーズであり、ポアを形成してもしなくてもよい。輸送方向は濃度勾配に従うこともあれば、従わないこともある(active,
または能動輸送)。Gating mechanismはない。Gating mechanismがないことは、たんぱく質の量、そして、それがどこにいるか、で輸送能力が決まることを意味する。
チャンネルたんぱく質をコードする遺伝子の変異によりさまざまな遺伝性疾患が生じることが明らかになっており、startle
diseaseもそのひとつである。まとめてチャンネル病と呼ばれるが、そのほとんどの表現型は直感的に理解できる。たとえば、水チャンネル
AQP2の機能欠損で尿崩症を生じ、ソディウムチャンネル SCN5 またはポタシウムチャンネル
HERG の機能欠損で Long QT syndrome (心電図のQT間隔が長くなり、不整脈を起こしやすくなる)を生じる。悪性高熱は麻酔科医の悪夢であり、骨格筋のライアノジン受容体の変異のため麻酔薬の副作用で筋壊死をきたす。嚢胞性肺繊維症
(cystic fibrosis: CF) はクロライドチャンネル CFTRの変異による。これは日本人には聞きなれない疾患だが、欧米では発症率の高いありふれた疾患であり、肺胞上皮細胞の機能障害により、呼吸器感染症を起こしやすくなり、肺が繊維化する。
同様に、トランスポーターをコードする遺伝子の変異によってもさまざまな遺伝性疾患が生じ、トランスポーター病と総称される。Startle
diseaseもそのひとつである。チャンネル病と同様、表現型はストレートに理解できるものが多い。Bartter症候群、Gitelman症候群はともに腎での電解質の輸送障害により血中の電解質平衡の異常をきたす。Glucose
transporter SGL1の欠損では消化管からの糖の吸収がうまくいかない。Tangier病ではABCA1の欠損により血管内皮細胞からHDLへのコレステロール・トランスポートがうまくいかず、低HDL血症と動脈硬化を生じる。ABCG5/G8の欠損では、胆汁・消化管へのコレステロール排出がうまくいかないために高脂血症になる。
これらに対して、中枢神経症状を呈すトランスポーター病の表現型を説明するのは難しい。Menkes病・Wison病では、銅のトランスポーターであるATP7A/ATP7Bの欠損のために神経症状を生じる。NPCでは細胞内でのコレステロールの輸送がうまくいかないために神経症状を呈す。これらの疾患において、なぜ銅、コレステロールの輸送異常が神経障害をきたすのかはわからない。
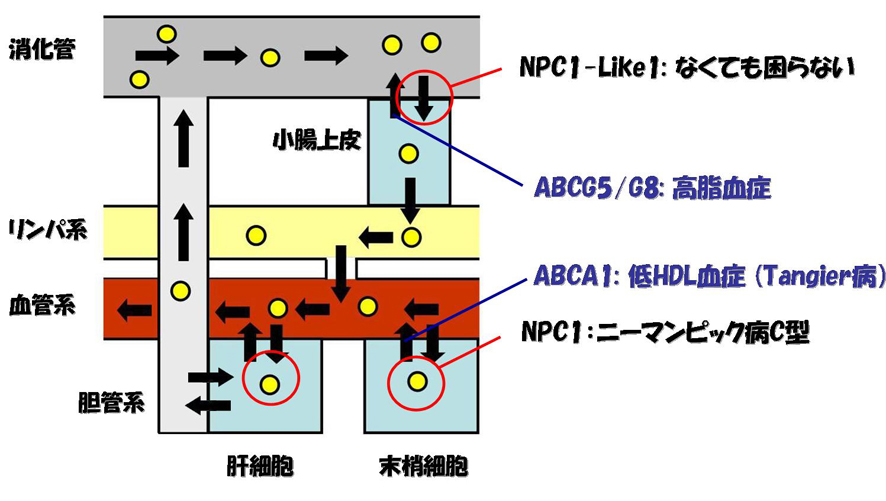
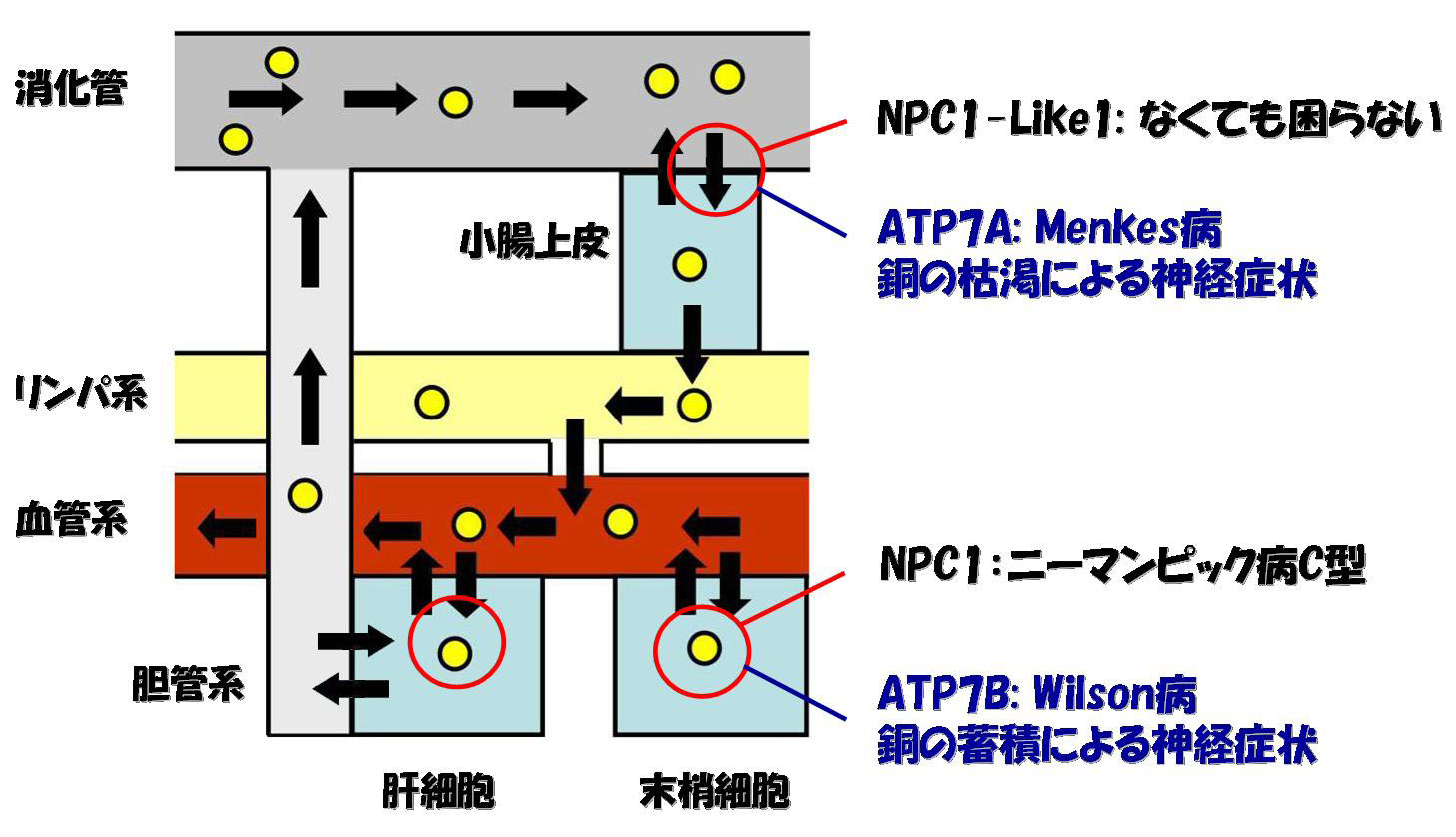
Note 1. ABCA1/ABCG5/G8とNPC1L1/NPC1
コレステロール・トランスポーターの局在と欠損のフェノタイプについてまとめておく。小腸上皮細胞では、消化管とコレステロールをやりとりする。入れるほうがNPC1-Like1であり、これは欠損しても何の症状もない。むしろこのトランスポーターが働かないことは、先進国の飽食の人々にとっては高脂血症・肥満の予防につながる。出すほうがABCG5/G8のヘテロダイマーであり、この欠損は高脂血症の原因になる。ヒトでは同じセットが肝細胞と胆管系の間でも同様に働いているらしい。
末梢細胞では、血管系とコレステロールをやりとりする。LDL受容体を介して入ってきたコレステロールを運搬するのがNPC1であり、この欠損はNPCである。HDLへコレステロールを排出するのがABCA1であり、この欠損はTangier病である。
|
コレステロール・トランスポーターの局在と欠損のフェノタイプ
|
ABCG5/G8の欠損による高脂血症では、動物性のステロールであるコレステロールよりは植物性のステロールであるphytosterolの血中の値が高くなる。これは、ABCG5/G8がphytosterolにより特異的なトランスポーターであることを意味する。
Note 2. ATP7A/ATP7BとNPC1L1/NPC
コレステロールと銅の輸送に関わるトランスポーターはその局在と欠損のフェノタイプに似たところがある。NPC1L1がコレステロールを体循環系に入れるのと同様に、銅を体内に入れるのがATP7Aであり、この欠損は銅の枯渇による神経疾患(Menkes病)をひきおこす。NPC1L1が無くてもこまらないのは、われわれがコレステロールを生合成できるからであり、ATP7Aが無いとまずいことになるのは銅を生合成できないからである。一方、NPC1が細胞内にはいってきたコレステロールを運搬するのと同様に、はいってきた銅を運搬するのがATP7Bであり、この欠損は銅の蓄積による神経疾患
(Wison病) をひきおこす。
|
NPC/NPC1L1によるコレステロール輸送と、ATP7A/ATP7Bによる銅の輸送
|
Phylogeneticalに考えると、消化管を持たない単細胞生物にはNPC1L1、ATP7Aは必要なかったはずで、これらはそれぞれNPC1、ATP7Bがduplicateしてできた遺伝子であると推察できる。小腸上皮細胞でこれらの遺伝子を使う理由は、その一次構造のなかに記述されているはずである。
Note 3. Menkes のATP7AとNPC1L1では、小腸上皮細胞のなかでの働きが異なる。Menkes病の小腸上皮細胞では、(他のすべての細胞とは逆に)銅が蓄積しており、ATP7Aは小腸上皮細胞の細胞内での輸送を担うとされている。これに対して、NPC1L1欠損の小腸上皮細胞ではコレステロールは枯渇している。
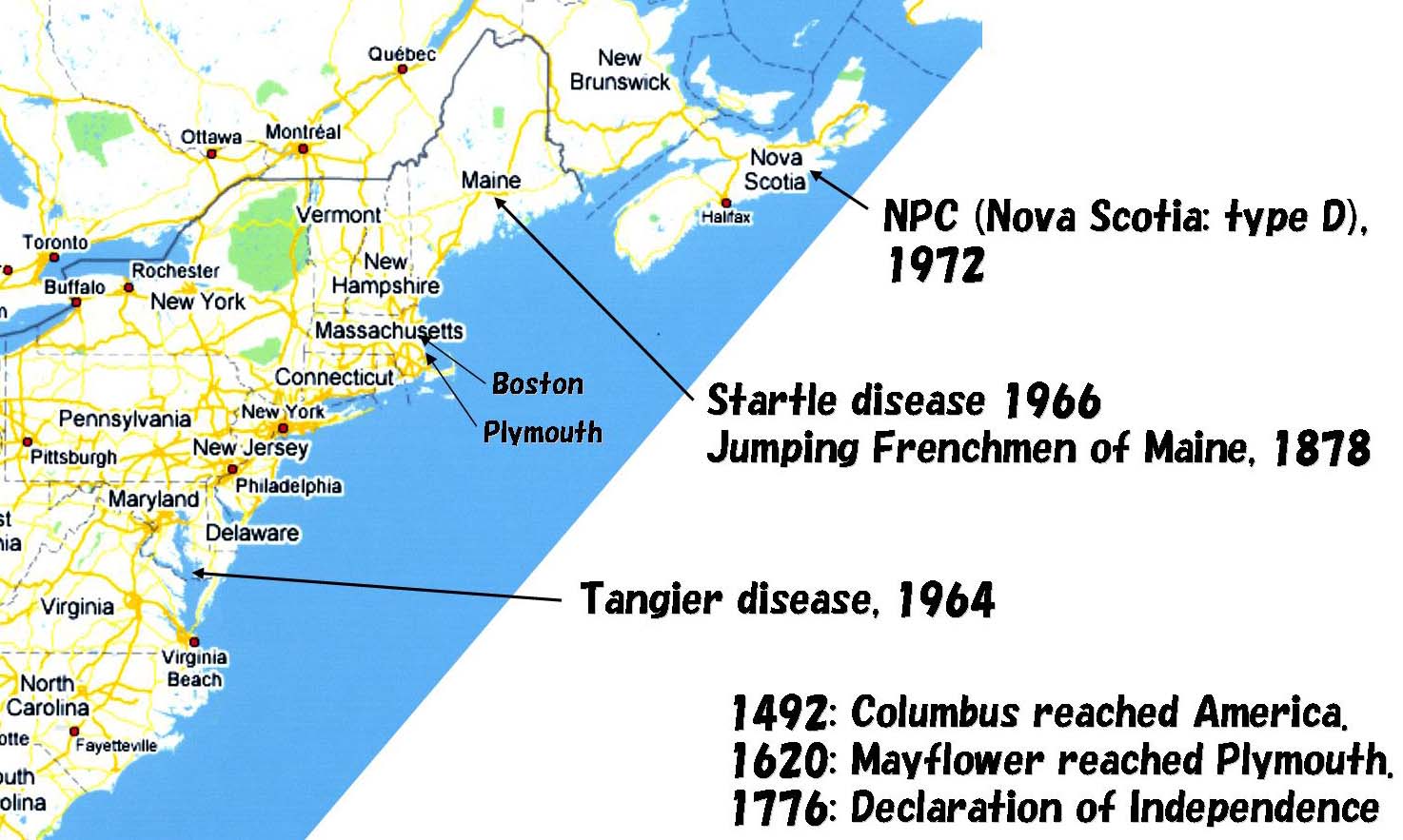
Tangier, Maine, Nova Scotia
このページに出てくる疾患の名前は、Wilson/Menkes/Niemann-Pickなど、たいていは最初にその疾患を記載した医者の名前に由来するが、Tangier病は例外であり、これは地名である。この病名は、その患者がアメリカ東海岸のTangier島に集積していることに由来する。1878年のstartle
diseaseの最初の記載は、アメリカ東海岸メイン州のある村に住むフランス人たちが驚いて飛び上がるという奇妙な行動を示したことを記している。古い教科書ではNiemann-Pick病の分類のひとつにNiemann-Pick
disease type D (NPD;Nova Scotia型)が記載されているが、これはメイン州の北、Nova
Scotia地方に患者が集積していることによる。現在では、NPDはNPC1の変異 G992W によることが判明しており、NPCに含まれている。NPC1
G992W について、1600年代にNova Scotiaに住んだフランス人夫婦Joseph MuiseとMarie
Amrout がともにこの変異のキャリアーだったことが判明しており、この地方の患者はすべてこのふたりのいずれかのアリールを継承している。
Tangier/startle disease/NPCのいずれも「非常にまれな」という枕言葉がつく遺伝性疾患だが、以上のことはこれらの「非常にまれな」疾患がわれわれに認識されるようになった歴史的背景を物語る。1492年にコロンブスがアメリカ大陸を発見した後、ヨーロッパからの移民が東海岸にやってくる。メイフラワー号がボストンの南の港町プリモスに着くのが1620年である。小船に乗ってわたってきた少人数の集団が、各々の新天地に定着し、世代を経てゆく。移動手段の発達していない時代にあって遺伝子プールの均質化が進み、各疾患の患者が集積することになる。
Tangier/startle disease/NPCは、1964, 66, 72年に各々遺伝性疾患としてカテゴライズされた。ダーウィンが「種の起源」を刊行したのは1859年である。その約100年後、1953年にワトソン&クリックがDNAの二重らせんモデルを発表する。21世紀の始めの今、私たちは、各々の疾患の原因遺伝子と、それが遺伝性疾患である理由を知っている。およそ疾患というものが自然の一部であり、それをコントロールすることが人類の目的であるならば、次の目標はこれらの疾患を治療することにある。それが夢で終わるのか、それとも、現実になる日がくるのか。
ページトップに戻る |